|
Lithium-ion batteries (LIBs) are the most commonly used energy source for household electronics and electric vehicles; however, these LIBs utilize a liquid electrolyte, which has a limited energy density. All-solid-state batteries, on the other hand, use a solid electrolyte – an inorganic, non-flammable ceramic material; thus, all-solid-state batteries have the potential to improve upon the energy density, and safety, of current Li-ion batteries.
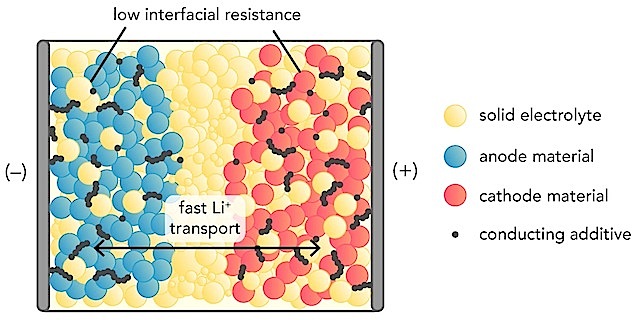
The development of all-solid-state batteries faces several challenges including a lack of fast Li-ion conducting ceramics, and mechanical and thermodynamic instability between solid ceramic interfaces. However, solid electrolytes and solid interfaces can be studied using computational materials modeling: a powerful tool used to investigate fundamental mechanisms, to predict and discover new materials, and to propose novel engineering strategies for all-solid-state batteries. Computation, for example, can predict new coatings for the interface at the anode, enabling lithium metal anodes, which provide significantly higher energy density than liquid electrolyte counterparts.
In the most recent issue of Joule, Yifei Mo – an assistant professor in the Department of Materials Science and Engineering (MSE) at the University of Maryland (UMD) – and his research group were invited to review state-of-the-art computational modeling techniques, and their application in the design of materials and interfaces for all-solid-state Li-ion batteries.
The review focuses on the unique contributions of computation to fundamental scientific understanding of solid-state battery materials at the atomic scale. Computation can predict the detailed dynamics of each individual atom on a femtosecond timescale (one thousandth of trillionth of a second).
“With computation, we can directly observe the diffusion mechanisms of lithium ions, which was impossible before,” said Adelaide Nolan, a MSE Ph.D. student and first author on the review. “Through these techniques, for the first time, we understand why some solid materials are fast ion conductors, which are critical to achieve high power in solid-state batteries.”
This understanding also laid the foundation for the discovery of new solid electrolyte materials with improved performance. First-principles computation is based on fundamental physical principles, allowing scientists to make predictions about new materials with no empirical input.
“The conventional way to develop new materials is very time consuming,” said Mo. “By using predictive quantum mechanical calculations combined with supercomputers, we can significantly accelerate the development of new battery materials such as solid electrolytes and electrodes.”
Dr. Mo’s group is a pioneer in this area – having discovered several new ion-conducting materials – and is working with industry partners to commercialize some of these computationally predicted materials. The research conducted by Mo’s group is performed on supercomputers at UMD.
This review was published online September 20th. For additional information:
Nolan, A.M.,Zhu, Y.,He, X.,Bai, Q.,Mo, Y. Computation-Accelerated Design of Materials and Interfaces for All-Solid-State Lithium-Ion Batteries. Joule, 20 Sept 2018. DOI: 10.1016/j.joule.2018.08.017.
Related Articles:
MSE Research Reveals Unique Ionic Diffusion Mechanism in Super-Ionic Conductors
Wachsman named Materials Research Society Fellow
UMD prominently showcased at ARPA-E summit
Former MEI2 energy seed grant discusses 3D printing of advanced ceramics
ION honored by federal and state officials
“Build Our Future” grants include two MEI2 energy companies
Climate Technology Founder's Fund (SB0960) to support new energy startups
Maryland’s first-ever solid-state battery pilot production line launches
$15M Federal Grant Awarded to Support Maryland Electric Vehicle Charging Network
Powering a Greener Future
September 24, 2018
|